
Ускладнення гемофілії
Гемофілія A — це виснажливе та небезпечне для життя захворювання.
Для людей, хворих на тяжку гемофілію A, очікувана тривалість життя зазвичай на 5 років менша, порівняно з людьми, які не мають гемофілії1. Хоча лікування гемофілії A з часом покращилося, зі збільшенням очікуваної тривалості життя, пацієнти, хворі на гемофілію A, особливо літньої вікової групи, усе ще страждають від серйозних ускладнень1,2.
Хронічні гемартрози є одними з найпоширеніших тривалих ускладнень гемофілії та, окрім захворюваності, пов’язані з пошкодженням суглобів; обмежена активність може призводити до остеопенії та ожиріння.
Інгібітори все ще залишаються найзначущішим ускладненням лікування гемофілії та майже у 30% людей, які хворіють на гемофілію A (PwHA), відбуватиметься розвиток інгібіторів (анти-FVIII антитіл) після введення екзогенного FVIII³.
Потреба в хорошому венозному доступі є критично важливою для пацієнтів, хворих на гемофілію. Отримати доступ до вен може бути складно, особливо в дітей молодшого віку, а повторювана венепункція може призвести до сильних кровотеч/утворення синців та незворотного пошкодження вен, що обмежує доступні ділянки введення лікарських засобів1,2.
Проривні кровотечі (кровотечі, які виникають у період профілактичного лікування пацієнта) залишаються проблемою з рівнями захисту, які пропонуються сучасними схемами профілактичного лікування з уведенням FVIII.
Повторні епізоди кровотеч мають негативні наслідки:

| *Порівняно з пацієнтами з гемофілією A без інгібіторів. |
|
У 1970-х і 1980-х роках, поряд із захворюваністю, спричиненою безпосередньо хворобою, багато пацієнтів, хворих на гемофілію A, були інфіковані ВГС або ВІЛ через заражену кров або препарати крові. І хоча поява концентратів рекомбінантних факторів згортання крові, вірусна інактивація, посилений скринінг та відсторонення від донорства призвели до того, що частота нових випадків інфікування була мінімальною з 1990-х років, загальна поширеність ВГС та ВІЛ у людей, хворих на гемофілію, залишається вищою, аніж у загальній популяції, з частотою 24–35% та 1,4–17% відповідно4-6.
Досягнення в розробці ефективних і безпечних методів лікування гемофілії за останні 50 років призвели до значного збільшення очікуваної тривалості життя осіб, хворих на гемофілію. Кількість багатьох ускладнень гемофілії, зокрема розвиток інгібіторів, внутрішньочерепний крововилив та ураження суглобів, збільшується з віком.
Життя з гемофілією A може серйозно вплинути на його якість, і пацієнти, хворі на гемофілію A, стикаються з низкою проблем на кожному етапі свого життя7.
З огляду на відсутність на роботі/у школі та оплату тривалого лікування, значним є, також, соціально-економічний тягар гемофілії8.
ЛІТЕРАТУРА
- Michael U. Callaghan, Claude Negrier, Ido Paz-Priel, Tiffany Chang, Sammy Chebon, Michaela Lehle, Johnny Mahlangu, Guy Young, Rebecca Kruse-Jarres, Maria Elisa Mancuso, Markus Niggli, Monet Howard, Nives Selak Bienz, Midori Shima, Victor Jiménez-Yuste, Christophe Schmitt, Elina Asikanius, Gallia G. Levy, Steven W. Pipe and Johannes Oldenburg «Long-term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1-4 studies», Blood. 2021 Apr 22; 137(16): 2231–2242. рrepublished online 2020 Dec 11. doi: 10.1182/blood.2020009217
- Young G. Hematology Am Soc Hematol Educ Program 2012;2012:362–368; 10Zhubi B et al. Bosn J Basic Med Sci 2009;9:271–7.
- Whelan SF et al. Blood 2013;121:1039–48.
- Zhubi B et al. Bosn J Basic Med Sci 2009;9:271–7.
- Northcott MJ et al. Haemophilia 2013;19:847–52.
- Forsyth AL et al. Haemophilia 2014;20:44–51.
- Cassis FR. Psychosocial care for people with hemophilia. Treatment of Haemophilia Monograph Series 2007. 44 http://www1.wfh.org/publications/files/pdf-1198.pdf.
- Caroll L et al. Haemophilia 2016;22 (Suppl. 4):38 (poster 60-PP-T).
ІНГІБІТОРИ
Лікування за допомогою екзогенного FVIII може призводити до виникнення нейтралізуючих анти-FVIII антитіл (інгібіторів), які зменшують ефективність лікування за допомогою FVIII шляхом перешкоджання зв’язуванню з тромбоцитами або іншими факторами згортання крові1. Інгібітори виникають у 25–30% пацієнтів, хворих на тяжку гемофілію A, та у 3–13% пацієнтів із помірною або легкою формою захворювання. Приблизно у 12–15% пацієнтів із тяжкою формою захворювання розвивається високий титр інгібіторів (≥5 одиниць Бетезда [ОБ]), унаслідок чого пацієнти перестають отримувати користь від терапії FVIII2–4.
Факторами ризику для розвитку інгібітора при гемофілії А є наявність інгібітора в сімейному анамнезі, належність до африканської та латиноамериканської раси, генетичні варіації (такі як тип мутації або поліморфні імунорегулюючі гени), а також високоінтенсивний вплив фактора (наприклад, інтенсивна замісна терапія при важкій кровотечі, що сталася раніше, крововилив у центральну нервову систему, хірургічній операції або при травмі)9.

Розвиток інгібіторів пов’язаний зі значним збільшенням захворюваності та смертності у людей, що страждають на гемофілію A (PwHA). Окрім зниження якості життя та підвищення вартості лікування, ведення кровотеч є складнішим та супроводжується більшою частотою ураження суглобів, порівняно з PwHA без розвитку інгібіторів5-7. У ретроспективному дослідженні людей, хворих на тяжку гемофілію A, що проводилося в США, наявність інгібіторів була пов’язана з більшою на 40% ймовірністю смерті, порівняно з людьми без інгібіторів, а також із більшою часткою смертей унаслідок кровотеч8.
Інгібітори слід запідозрити при кровотечах, що продовжуються, попри достатню замісну терапію із введенням фактора. Наявність нового інгібітора слід запідозрити у будь-якого пацієнта, у якого немає клінічної відповіді на фактори згортання, особливо якщо така відповідь раніше була9.
При тяжкій гемофілії, наявність інгібіторів не впливає на локалізацію, частоту або ступінь тяжкості кровотечі. При помірній або легкій гемофілії інгібітор може нейтралізувати ендогенно синтезований FVIII та, у такий спосіб, перетворювати фенотиповий прояв захворювання пацієнта на тяжку форму.
Дітям скринінг на інгібітори слід проводити один раз кожні п’ять днів лікування фактором протягом 20 днів лікування, кожні 10 днів лікування фактором між 21 і 50 днем лікування, і, принаймні, два рази на рік протягом 150 днів лікування.
Для дорослих, які понад 150 днів отримували лікування фактором, окрім 6–12-місячної оцінки, будь-яка відсутність реакції на замісну терапію концентратів адекватного фактора у пацієнтів, які раніше мали реакції, є показанням для проведення оцінки на наявність інгібітора.
Вимірювання інгібітора слід також проводити у всіх пацієнтів, які інтенсивно лікувалися понад п’ять днів, протягом чотирьох тижнів із моменту останнього вливання.
Наявність інгібіторів також має бути оцінена до проведення хірургічного втручання або якщо результати аналізу відновлення не відповідають очікуваним, а також коли клінічна відповідь на лікування недостатня в післяопераційний період9.
Пацієнти, хворі на легку форму гемофілії A, а також пацієнти, які вперше отримують інтенсивну замісну терапію з введенням фактора, мають певний ризик розвитку інгібітора та повинні пройти повторний скринінг через 4–12 тижнів після проведення процедури.
Також рекомендується проводити ретельний моніторинг на наявність інгібіторів у пацієнтів із нетяжкою формою гемофілії A, які отримують неперервну інфузію після хірургічного втручання.
Передопераційна оцінка має включати скринінг на наявність та кількість інгібітора, особливо якщо рівень відновлення заміщеного фактора суттєво менший за очікуваний.
Інгібітори вимірюються за допомогою аналізу Бетезда або аналізу Бетезда в модифікації Ніймеген. Позитивною відповіддю на наявність інгібітора є титр Бетезда величиною >0,6 одиниць Бетезда (БЕ) для FVIII та ≥0,3 БЕ для FIX9.
При високореагуючих інгібіторах для лікування кровотеч застосовують препарати з шунтуючим механізмом дії (рекомбінантний активований фактор VIIa [rFVIIa] або активований концентрат протромбінового комплексу [аКПК]) або свинячий FVIII. У оновленому 3-му виданні Рекомендацій WFH, щодо лікування пацієнтів з гемофілією, зазначається, що еміцизумаб слід застосовувати для регулярної профілактики у пацієнтів з Гемофілією А з наявністю інгібіторів9
ЛІТЕРАТУРА
- Whelan SF et al. Blood 2013;121:1039–48.
- Wight J and Paisley S. Haemophilia 2003;9:418–35.
- Gouw SC et al. Blood 2007; 109: 4648–54.
- Eckhardt CL et al. Blood 2013;122:1954–62.
- Gringeri A et al. Blood 2003;102: 2358–63.
- Morfini M et al. Haemophilia 2007;13:606–12.
- Recht M, et al. Value Health. 2014;17:744–8.
- Walsh CE et al. Am J Hematol 2015;90:400–5.
- WFH Guidelines for the management of hemophilia, 3-d edition, https://elearning.wfh.org/resource/treatment-guidelines/ доступ, травень 2023
ГЕМОФІЛЬНА АРТРОПАТІЯ
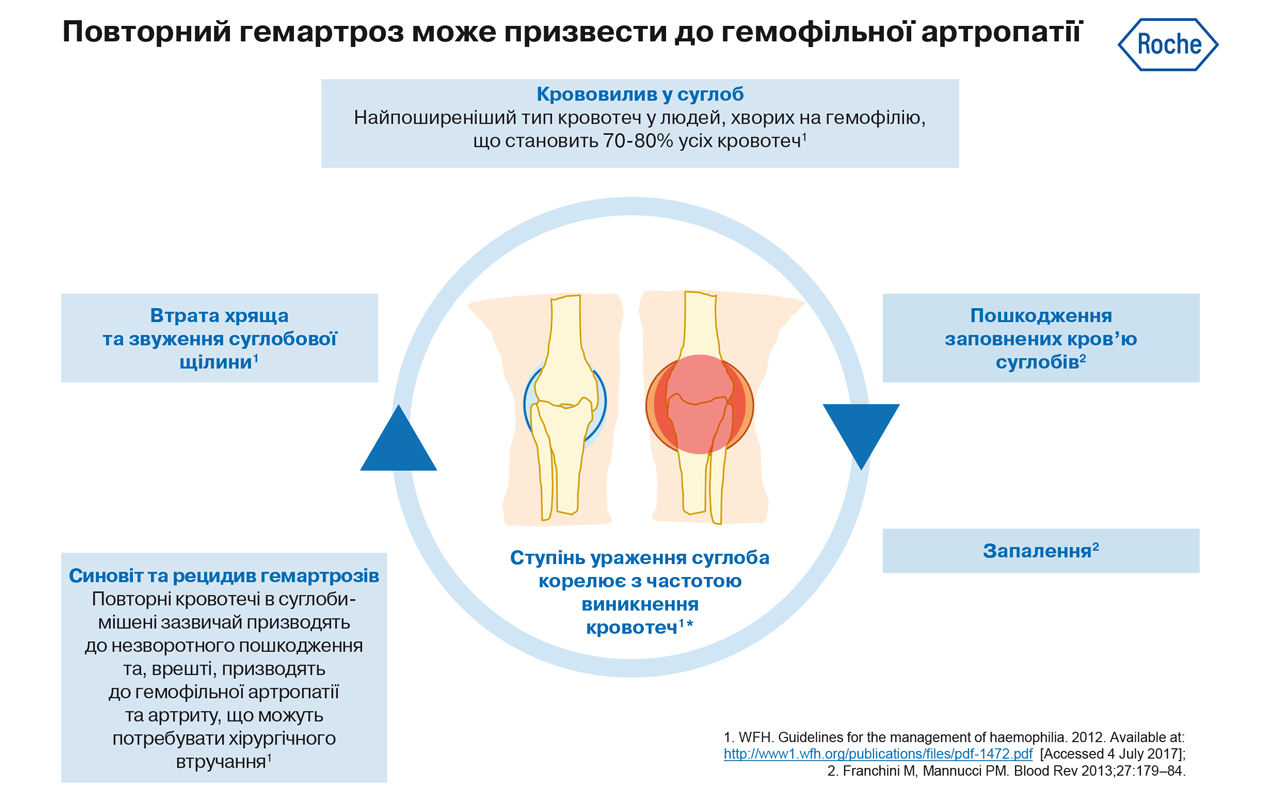
Близько 90% епізодів кровотечі у пацієнтів з гемофілією трапляються в опорно-руховому апараті, і з них 80% в суглоби. Найчастіше уражаються колінні, гомілковостопні та ліктьові суглоби. Наслідки рецидивуючих кровотеч в суглоб - хронічний синовіт та руйнування суглобового хряща та субхондральної кістки. Цей стан, який називається хронічною гемофільною артропатією, викликає біль, жорсткість та деформацію, що призводить до сильного порушення функції ураженого суглоба. Ураження колінного суглоба, пов'язане з найбільш значною інвалідизацією та погіршенням якості життя, а повна заміна колінного суглобу, є найпоширенішою операцією по загальній заміні суглобів, що проводиться у людей з гемофілією1,2.
Ранніми клінічними ознаками гемартрозу є підвищене відчуття тепла або «аура» над ураженою ділянкою та дискомфорт при русі, що супроводжується болем, запаленням та вкрай обмеженою рухливістю4.
Суглоби, у яких виникають повторні кровотечі (зазвичай три або більше протягом 6-місячного періоду), класифікуються як суглоби-мішені.
Повторні кровотечі в суглоби-мішені зазвичай спричиняють незворотне пошкодження суглоба, що врешті призводить до гемофільної артропатії та артриту. Запалення внаслідок повторних кровотеч може призвести до синовіту, руйнування хряща та звуження суглобової щілини, причому ступінь ураження суглоба корелює з кількістю кровотеч.
Гострі гемартрози, як правило, починаються з легкого дискомфорту та незначного обмеження рухів у суглобі, що супроводжуються болем, набряком суглобів та відчуттям тепла на шкірі. За відсутності лікування крововилив у суглоб призводить до вираженого обмеження рухів.
На жаль, патологічні процеси тривають навіть після зупинки кровотечі, оскільки запалення спричиняє пошкодження заповнених кров’ю суглобів, яке призводить до синовіту, що, зі свого боку, збільшує ймовірність частих рецидивів кровотеч у тих самих суглобах (так звані суглоби-мішені).
Останнім етапом цього хибного кола, який спричиняє гемофільну артропатію, є звуження суглобової щілини через втрату хряща, розвиток кісткових кіст та обмеження рухів, що призводить до стійкої втрати працездатності4.
Рентгенологічні методи для оцінки хронічної гемофілічної артропатії вибираються згідно стадії прогресуючого перебігу захворювання.
МРТ доцільно використовуватиме оцінки початкової артропатії та для візуалізації ранніх змін у м'яких тканинах та кістково-хрящових структурах.
Ультразвукове дослідження допомагає оцінити патологію в м'яких тканинах та по периферії хряща на ранній стадії гемофілічної артропатії.
Звичайна рентгенограма нечутлива до ранніх змін і використовується для оцінки артропатичних змін на пізніх стадіях3.
Сучасні схеми профілактики із введенням концентратів фактора згортання є ефективними, хоча вони не запобігають розвитку артропатії суглоба протягом усього життя.
Поява артропатії суглоба переважно залежить від кількості кровотеч на рік, особливо гемартрозів на рік, які стали сурогатними маркерами результатів як у клінічних, так і післяреєстраційних дослідженнях. Хоча у пацієнтів, які отримують лікування за потребою, відзначається від 20 до 50 кровотеч на рік та розвиток артропатії суглобів у ранньому віці, у пацієнтів, які отримують профілактичне лікування, може не бути проявів ураження суглобів протягом 1–2 десятиліть або навіть довше5.
До настання ери профілактики найчастіше серед гемартрозів відзначалися кровотечі в колінному суглобі. У більшості пацієнтів колінні суглоби посідали головне місце серед клінічних проявів. З початком ери профілактики колінний суглоб, як контрольований м’язами суглоб, став стійкішим, а суглоби щиколотки стали суглобами, які вражаються найпершими, і тому представляють клінічно найхарактерніший суглоб. Щиколотки, як перші уражені суглоби для більшості пацієнтів, є цікавими з точки зору діагностики ранніх уражень суглобів та подальшого прийняття клінічних рішень.
Стандартом медичної допомоги вважається профілактичне лікування, спрямоване на запобігання геморагічних епізодів6.
Ціль лікування полягає в тому, щоб зменшити частоту гемартрозів, покращити функціонування суглобів, зменшити біль та допомогти пацієнтові продовжити або відновити виконання звичайних для повсякденного життя дій.
Варіанти лікування хронічної гемофілічної артропатії залежать від багатьох факторів, зокрема: стадії у патологічному процесі; симптомів пацієнта; віку пацієнта; впливу на стиль життя пацієнта та на його функціональні можливості; наявних ресурсів3.
Для контролю больового синдрому, що викликається хронічною гемофілічною артропатією,у дітей та дорослих з гемофілією та болем, ВФГ рекомендує використовувати парацетамол/ацетамінофен, селективні інгібітори ЦОГ-2, трамадол або морфін; інших нестероїдних протизапальних засобів слід уникати. Кодеїн можна використовувати для дітей віком від 12 років, дітям молодшого віку він протипоказаний. При цьому зазначаэться, що тривалий прийом цих медикаментів несе у собі ризик звикання та залежності, а також ушкодження органів, тому потрібне уважне спостереження3.
Одним із пріоритетів у покращенні стану здоров’я та якості життя людей, хворих на гемофілію, є профілактика кровотеч та пошкодження суглобів. Профілактика не допоможе відновити суглоби, які вже пошкоджені. Однак вона зменшить частоту кровотеч, може уповільнити прогресування ураження суглобів та покращити якість життя людей, хворих на гемофілію A3.
ЛІТЕРАТУРА
- Jerome Wiedel, Sally Stabler, Sharon Funk “JOINT REPLACEMENT SURGERY IN HEMOPHILIA” https://www1.wfh.org/publication/files/pdf-1210.pdf
- E. Carlos Rodríguez-Merchán “The role of orthopaedic surgery in haemophilia: current rationale, indications and results”, EFORT open review, Volume 4: Issue 5, p. 165–173 https://eor.bioscientifica.com/view/journals/eor/4/5/2058-5241.4.180090.xml
- WFH Guidelines for the management of hemophilia, 3-d edition, https://elearning.wfh.org/resource/treatment-guidelines/ доступ, травень 2023
- Franchini M and Mannucci PM. Blood Rev 2013;27:179–84.
- Oldenburg J. Blood (2015) 125 (13): 2038–2044.
- Poonnoose P, Carneiro JDA, Cruickshank AL, et al. Episodic replacement of clotting factor concentrates does not prevent bleeding or musculoskeletal damage—the MUSFIH study. Haemophilia.2017;23(4):538-546
УТРУДНЕНИЙ ВЕНОЗНИЙ ДОСТУП
Потреба в хорошому венозному доступі є критично важливою для пацієнтів, хворих на гемофілію. Отримати доступ до вен може бути складно, особливо в дітей молодшого віку, а повторювана венепункція може призвести до сильних кровотеч/утворення синців та незворотного пошкодження вен, що обмежує доступні ділянки введення лікарських засобів1,2.
Утруднений венозний доступ (DIVA, difficult venous access), часто зустрічається в медичній практиці. За попередньою оцінкою, більше половини всіх, госпіталізованих, пацієнтів, потребують встановлення периферичного внутрішньовенного катетера (ПВК) для парентерального введення ліків. Це найпоширеніша інвазійна клінічна процедура. Понад третина дорослих і до половини дітей, які звертаються до лікарні та потребують ПВК, мають утруднений венозний доступ. Для визначення утрудненого венозного доступу, використовуються різні підходи, засновані на статі, вікові пацієнтів та багатьох інших параметрах. Широко вживаною, у дорослих пацієнтів, є шкала, для визначення факторів ризику розвитку утрудненого венозного доступу А-DIVA, яка включає декілька критеріїв: кількість периферичних вен, які можно визначити візуально; кількість периферичних вен, що пальпуються; наявність, в анамнезі невдалих спроб проведення внутрішньовенних інфузій; позапланове проведення оперативних втручань; діаметр вени ≤2 мм. При оцінці за цією шкалою, якщо сума балів ≥ 4, то такий випадок, трактується як утруднений венозний доступ3.
Також за даними різних публікацій до факторів ризику утрудненого венозного доступу, відноситься індекс маси тіла (ІМТ ≥ 30 або ІМТ < 18)4.
ЛІТЕРАТУРА
- Mauser-Bunschoten EP et al. Haemophilia 2009;15:853–63.
- Young G. Hematology Am Soc Hematol Educ Program 2012;2012:362–368; 10Zhubi B et al. Bosn J Basic Med Sci 2009;9:271–7.
- Vanno Sou et all “A clinical pathway for the management of difficult venous access” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5693534/#CR5
- Michael U. Callaghan, Claude Negrier, Ido Paz-Priel, Tiffany Chang, Sammy Chebon, Michaela Lehle, Johnny Mahlangu, Guy Young, Rebecca Kruse-Jarres, Maria Elisa Mancuso, Markus Niggli, Monet Howard, Nives Selak Bienz, Midori Shima, Victor Jiménez-Yuste, Christophe Schmitt, Elina Asikanius, Gallia G. Levy, Steven W. Pipe and Johannes Oldenburg «Long-term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1-4 studies», Blood. 2021 Apr 22; 137(16): 2231–2242. рrepublished online 2020 Dec 11. doi: 10.1182/blood.2020009217
- Інструкція для медичного застосування лікарського засобу Гемлібра®затвержена Наказом МОЗ України №1572 від 30.08.2018, зміни внесено Наказом МОЗ України №813 від 16.05.2022 Реєстраційне посвідчення МОЗ України UA/16914/01/02, UA/16914/01/01.